SLUSCHI

Documentation
This work is licensed under a Creative Commons Attribution-NoDerivatives 4.0 International License.
Please contact me at [email protected] if you have questions.
Several Notes for Users:
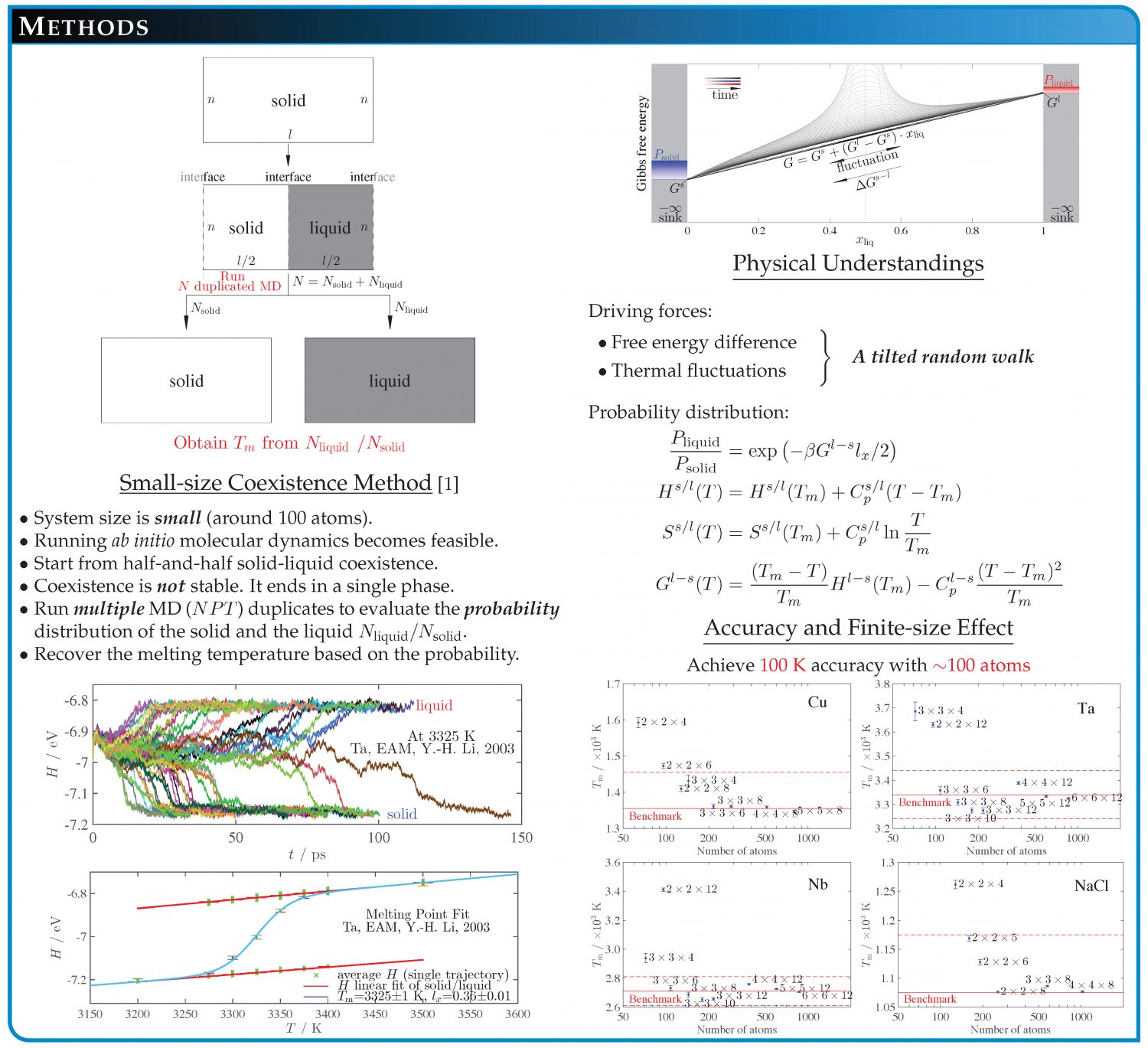
- A typical calculation with SLUSCHI takes a few days/weeks, with CPU cost ranging from 5,000 to 100,000 CPU hours. On this page, I list the systems I have studied.
- Try “kmesh = -1” in “job.in”. This activates the use of a special kpoint (1/4,1/4,1/4). This provides a low-cost option (only twice the cost of gamma version), and yet it often generates reliable results for melting point.
- An example of SLUSCHI run is available at the repositories at the National Institute of Standards and Technology. Or follow this link on Google Drive for the same copy.
Sluschi_2.0 Github
What’s new:
- mds: a new capability to compute the entropy of both solids and liquids from one single MD trajectory. A user manual is coming soon this fall. Follow this link on Google Drive for examples.
- Bug fixes and various improvements.
Sluschi_1.3
What’s new:
- Add feature to detect failed VASP jobs and restart them. It takes hundreds of VASP MD runs to calculate a melting temperature, and some of them may accidentally fail (e.g., running beyond walltime limit, failed to start due to queue/disk/network issue, etc.). Now SLUSCHI checks VASP MD runs and restarts failed jobs. Set “detectfail” and “maxwaithour” in job.in.
- Bug fixes and various improvements.
Sluschi_1.2
What’s new:
- Bug fixes and various improvements.
Sluschi_1.1
What’s new:
- “Bold sampling” mode: SLUSCHI launches MD duplicates aggressively. This strategy significantly reduces the physical time of calculations, though it may slightly increase computer cost.
- Heat of fusion calculations
- Bug fixes and various improvements.
Sluschi_1
User guide at the CALPHAD Journal.
What’s new:
- intpol: user does not need to guess a melting point. See “intpol”.
- adj_bmix: let SLUSCHI decide the value of BMIX.
- Bug fixes and various improvements.
Download Counts by 2021
Traffic analyzed at BrownBox by Brown University, before I left Brown and moved SLUSCHI to GitHub.

What is SLUSCHI
SLUSCHI is a fully automated code which calculates melting points based on first-principles molecular dynamics simulations, with interface to the first-principles code VASP. Starting from the crystal structure of a solid (which the user inputs), SLUSCHI will automatically build a supercell of a proper size, prepare solid-liquid coexistence, and then employ the small-cell coexistence method to calculate the melting temperature. SLUSCHI is applicable to a wide variety of materials, thanks to the fact that density functional theory calculations are highly generalizable.
Systems Tested
Ta (0 and 200 GPa, bcc), Na (30-120 GPa, bcc/fcc), NaCl, La2Zr2O7 (La2O3-2ZrO2), the Hf-Ta-C-N systems, Al, Si, HfO2, Fe (330 GPa), Ti (bcc) and many more…
m.p.: melting point calculated by SLUSCHI.
If not specified, PAW-PBE is employed in the calculations.
HSE: melting point after HSE correction. (important for semiconductors)
MD: total number of MD trajectories
CPU hours are measured as / converted to TACC Stampede.
days: the physical time it takes. SLUSCHI is currently optimized to lower the CPU cost, rather the physical time. In order to reduce the physical time, user may manually explore temperature of interest. However, this may increase CPU hours.
“early runs” are calculations performed at early stage of method development. Hence the efficiency is relatively low compared to “sluschi”.
poor results and reason | good results
| systems | m.p./K | HSE/K | expt./K | DFT PP | rad / Å | kmesh | # MD | cpu hours | days | note |
| Al | 1040±13 | 933 | Al | 12 | (1/2,1/2,1/2) | 19 | 5,400 | 7 | sluschi | |
| Al | 999±21 | 1054 | 933 | Al | 10 | Auto 30 | 58 | 16,000 | 15 | sluschi |
| Ti_v | 1811±47 | 1941 | Ti_v | 9 | Auto 20 | 49 | 15,500 | 21 | sluschi | |
| Ti_v | 1750±25 | 1971 | 1941 | Ti_v | 10 | Auto 20 | 26 | 7,900 | 17 | sluschi |
| Ti_pv | 1952±45 | 1941 | Ti_pv | 10 | (1/4,1/4,1/4) | 48 | 20,000 | 20 | sluschi | |
| Ti_pv 60GPa | 2505±47 | ? | Ti_pv | 10 | (1/4,1/4,1/4) | 46 | 39,500 | 17 | sluschi | |
| Si | 724±47 | 1687 | Si | 10 | Auto 10 | 25 | 1,000 | 2 | sluschi | |
| Si | 1378±24 | 1785 | 1687 | Si | 10 | Auto 20 | 19 | 7,500 | 15 | el. DOS change, require HSE |
| Si | 1364±37 | 1687 | Si | 12 | Auto 20 | 20 | 27,700 | 27 | sluschi | |
| diamond,100GPa | 4307±8 | 4250 | C | 10 | gamma | 45 | 313,000 | 168 | sluschi | |
| Ru_v | 2435±32 | 2607 | Ru_v | 10 | Auto 20 | 30 | 88,000 | 37 | sluschi | |
| Ru_pv | 2550±34 | 2607 | Ru_pv | 10 | (1/4,1/4,1/4) | 33 | 139,000 | 51 | sluschi | |
| Ru ternary alloys | 2564±40 | n/a | pv | 10 | (1/4,1/4,1/4) | 23 | 247,000 | 80 | sluschi | |
| Hf,bcc | 2562±31 | 2506 | Hf | 10 | (1/2,1/2,1/2) | 115 | 14,900 | 12 | sluschi | |
| Hf,hcp | 2122±50 | n/a | Hf | 10 | (1/2,1/2,1/2) | 50 | 25,800 | 21 | sluschi, hcp not stable at MT | |
| HfO2 | 2327±47 | – | 3031 | valence | 10 | gamma | 33 | 79,000 | sluschi | |
| HfO2 PBE+U | 3486±91 | – | 3031 | Hf_pv | 10 | gamma | 34 | 99,600 | sluschi | |
| HfO2 PBE+U | 3313±81 | – | 3031 | Hf_pv | 12 | gamma | 25 | running | ionic, use large $rad | |
| ZrO2 | 2988 | valence | 10 | Auto 20 | running | |||||
| Ta_v | 2986±41 | 3290 | Ta_v | 10 | (0,1/4,1/4) | 38 | 32,000 | 23 | early runs, low efficiency | |
| Ta_pv | 3194±40 | 3290 | Ta_pv | 10 | (0,1/4,1/4) | 38 | 54,000 | 24 | early runs, low efficiency | |
| Ta_pv_PW91 | 3066±51 | 3290 | Ta_pv | 10 | (0,1/4,1/4) | 38 | 38,000 | 68 | PW91 [1] | |
| Ta_pv,200GPa | 7953±69 | n/a | Ta_pv | 10 | (1/4,1/4,1/4) | 80 | 150,000 | 48 | sluschi, high efficiency [4] | |
| W | 3497±54 | 3695 | W | 10 | A20 | 22 | 35,900 | 49 | sluschi | |
| W_pv | 3470±45 | 3695 | W_pv | 10 | (1/4,1/4,1/4) | 30 | 38,500 | 18 | sluschi | |
| Na 15 GPa | 657±8 | 810, 698 ? | Na_pv | 10.4 | (0,1/4,1/4) | 55 | 47,000 | 24 | bcc, expt under dispute, e.g., | |
| Na 26 GPa | 750±16 | 706 | 970 ? | Na_pv | 9.8 | (0,1/4,1/4) | 52 | 26,000 | 17 | Zha, Boehler vs. Gregoryanz |
| Na 40 GPa | 742±17 | 950 ? | Na_pv | 9.4 | (0,1/4,1/4) | 74 | 54,000 | 37 | SLUSCHI results agree with | |
| Na 55 GPa | 716±12 | 810 ? | Na_pv | 9.0 | (0,1/4,1/4) | 56 | 64,000 | 31 | Eshet and Desjarlais (theory). | |
| Na 80 GPa | 674±20 | 700 ? | Na_pv | 10.9 | (0,1/4,1/4) | 55 | 170,000 | 67 | fcc, expt under dispute | |
| Na 100 GPa | 662±14 | 450 ? | Na_pv | 10.6 | (0,1/4,1/4) | 48 | 57,000 | 28 | fcc, expt under dispute | |
| Na 120 GPa | 579±27 | ? | Na_pv | 10.4 | (0,1/4,1/4) | 88 | 150,000 | 77 | fcc, expt under dispute | |
| NaCl | 1014±18 | 1074 | valence | 11 | gamma | 59 | 24,000 | 50 | early runs, low efficiency | |
| La2Zr2O7 | 2420±27 | 2630 | 2530 | Zr_v, La_sv | 10 | gamma | 64 | 575,000 | 210 | [2] |
| Hf-Ta-C-N | – | – | valence | 10 | – | – | – | [3] | ||
| ⋮ |
- Qi-Jun Hong and Axel van de Walle, Solid-liquid coexistence in small systems: A statistical method to calculate melting temperatures. Journal of Chemical Physics 139 (9), 094114 (2013). [DOI]
- Qi-Jun Hong, Sergey V. Ushakov, Alexandra Navrotsky, Axel van de Walle, Combined computational and experimental investigation of the refractory properties of La2Zr2O7. Acta Materialia 84, 275-282 (2015). [DOI]
- Qi-Jun Hong and Axel van de Walle, Prediction of the material with highest melting temperature from quantum mechanics. Physical Review B Rapid Communications, 92, 020104(R) (2015). [DOI]
- Ljubomir Miljacic, Steven Demers, Qi-Jun Hong and Axel van de Walle, Equation of state of solid, liquid and gaseous tantalum from first principles. Calphad: Computer Coupling of Phase Diagrams and Thermochemistry, 51, 133-143 (2015). [DOI (open access)].